|
The
past few years have brought some encouraging studies to the
forefront in Alzheimer's and Dementia with several drugs in
the pipeline that show promise
BIOLOGICAL BASIS
Three
major competing hypotheses exist to explain the cause of the
disease. The oldest, on which most currently available drug
therapies are based, is the cholinergic hypothesis, which proposes
that AD is caused by reduced synthesis of the neurotransmitter
acetylcholine. The cholinergic hypothesis has not maintained
widespread support, largely because medications intended to
treat acetylcholine deficiency have not been very effective.
1)
The cholinergic hypothesis, which proposes that AD is
caused by reduced synthesis of the neurotransmitter acetylcholine.
The cholinergic hypothesis has not maintained widespread support,
largely because medications intended to treat acetylcholine
deficiency have not been very effective. The cholinergic hypothesis
is the oldest hypothesis and is what most current drug therapies
are based upon.Other cholinergic effects have also been proposed,
for example, initiation of large-scale aggregation of amyloid,[1]
leading to generalised neuroinflammation [2].
2)
In 1991, the amyloid hypothesis postulated that amyloid
beta (Aß) deposits are the fundamental cause of the disease.[3][4]
Support for this postulate comes from the location of the gene
for the amyloid beta precursor protein (APP) on chromosome 21,
together with the fact that people with trisomy 21 (Down Syndrome)
who thus have an extra gene copy almost universally exhibit
AD by 40 years of age.[5][6] Also APOE4, the major genetic
risk factor for AD, leads to excess amyloid buildup in the brain
before AD symptoms arise. Thus, Aß deposition precedes clinical
AD.[43] Further evidence comes from the finding that transgenic
mice that express a mutant form of the human APP gene develop
fibrillar amyloid plaques and Alzheimer's-like brain pathology
with spatial learning deficits.[7]
--Note:
An experimental vaccine however was found to clear the amyloid
plaques in early human trials, but it did not have any significant
effect on dementia.[8] --
Researchers
have been led to suspect non-plaque Aß oligomers (aggregates
of many monomers) as the primary pathogenic form of Aß. In 2009,
it was found that oligomeric Aß exerts a deleterious effect
on brain physiology by binding to a specific receptor on neurons.
The identity of this receptor is the prion protein that has
been linked to mad cow disease and the related human condition,
Creutzfeldt-Jakob disease, thus potentially linking the underlying
mechanism of these neurodegenerative disorders with that of
Alzheimer's disease.[9].
In
2009, this theory was updated, suggesting that a close relative
of the beta-amyloid protein, and not necessarily the beta-amyloid
itself, may be involved in the disease (see Ref 10). The theory
holds that an amyloid-related mechanism that prunes neuronal
connections in the brain in the fast-growth phase of early life
may be triggered by aging-related processes in later life to
cause the neuronal withering of Alzheimer's disease. N-APP,
a fragment of APP from the peptide's N-terminus, is adjacent
to beta-amyloid and is cleaved from APP by one of the same enzymes.
N-APP triggers the self-destruct pathway by binding to a neuronal
receptor called death receptor 6 (DR6, also known as TNFRSF21).[10]
DR6 is highly expressed in the human brain regions most affected
by Alzheimer's, so it is possible that the N-APP/DR6 pathway
might be expressed in the aging brain to cause damage. In this
model, Beta-amyloid plays a complementary role, by depressing
synaptic function.
3)
A 2004 study found that deposition of amyloid plaques does not
correlate well with neuron loss.[11] This observation supports
the tau hypothesis, the idea that tau protein abnormalities
initiate the disease cascade. Tau proteins are microtubule-associated
proteins that are abundant in neurons in the central nervous
system and are less common elsewhere. In
this model, hyperphosphorylated tau begins to pair with other
threads of tau. Eventually, they form neurofibrillary tangles
inside nerve cell bodies.[12] When this occurs, the microtubules
disintegrate, collapsing the neuron's transport system.[13]
This may result first in malfunctions in biochemical communication
between neurons and later in the death of the cells.[14] Herpes
simplex virus type 1 has also been proposed to play a causative
role in people carrying the susceptible versions of the apoE
gene.[15]
Biochemistry
Enzymes
act on the APP (amyloid precursor protein) and cut it into fragments.
The beta-amyloid fragment is crucial in the formation of senile
plaques in AD. Alzheimer's disease has been identified as a
protein misfolding disease (proteopathy), caused by accumulation
of abnormally folded A-beta and tau proteins in the brain. Plaques
are made up of small peptides, 39–43 amino acids in length,
called beta-amyloid . Beta-amyloid is a fragment from a larger
protein called amyloid precursor protein (APP), a transmembrane
protein that penetrates through the neuron's membrane. APP is
critical to neuron growth, survival and post-injury repair.
In Alzheimer's disease, an unknown process causes APP to be
divided into smaller fragments by enzymes through proteolysis.
One of these fragments gives rise to fibrils of beta-amyloid,
which form clumps that deposit outside neurons in dense formations
known as senile plaques.
AD
is also considered a tauopathy due to abnormal aggregation of
the tau protein. Every neuron has a cytoskeleton, an internal
support structure partly made up of structures called microtubules.
These microtubules act like tracks, guiding nutrients and molecules
from the body of the cell to the ends of the axon and back.
A protein called tau stabilises the microtubules when phosphorylated,
and is therefore called a microtubule-associated protein. In
AD, tau undergoes chemical changes, becoming hyperphosphorylated;
it then begins to pair with other threads, creating neurofibrillary
tangles and disintegrating the neuron's transport system.
Genetic
Predisposition to Alzheimer's
There are
two categories of genes that can play a role in determining
whether a person develops a disease. Alzheimer genes have been
found in both categories:
1) Risk
genes increase the likelihood of developing a disease, but
do not guarantee it will happen.
Scientists
have so far identified one Alzheimer risk gene called apolipoprotein
E-e4 (APOE-e4).APOE located on chromosome 19, contains the instructions
needed to make a protein (apolipoprotein E) that helps carry
cholesterol in the bloodstream. APOE comes in several different
forms, or alleles. Three forms—APOE e2, APOE e3, and APOE e4—occur
most frequently. APOE e2 is relatively rare and may provide
some protection against the disease. If AD does occur in a person
with this allele, it develops later in life than it would in
someone with the APOE e4 gene. APOE e3 is the most common allele.
Researchers think it plays a neutral role in AD—neither decreasing
nor increasing risk. APOE e4 occurs in about 40 percent
of all people who develop late-onset AD and is present in about
25 to 30 percent of the population. People with AD are more
likely to have an APOE e4 allele than people who do not develop
AD. People who inherit one copy of the APOE e4 allele have an
increased chance of developing the disease; those who inherit
two copies of the allele are at even greater risk. It is not
known how the APOE e4 allele is related to the risk of Alzheimer
disease. Researchers have found that this allele is associated
with an increased number of protein clumps, called amyloid plaques,
in the brain tissue of affected people. It must however be pointed
out that many people with AD do not have an APOE e4 allele.
|
NOTE:
While the exact mechanism of how E4 causes such dramatic
effects remains to be fully determined, evidence has been
presented suggesting an interaction with amyloid. Alzheimer's
Disease is characterized by plaques consisting of the
peptide beta-amyloid. Apolipoprotein E enhances proteolytic
break-down of this peptide, both within and between cells.
Some isoforms of ApoE are not as efficient as others at
catalyzing these reactions. In particular, the isoform
ApoE-Ee4 is not very effective, resulting in increased
vulnerability to Alzheimer's in individuals with that
gene variation. See
also: ApoE
and Alzheimer's Disease — Structure of APOE
|
2) Deterministic
genes directly cause a disease, guaranteeing that anyone
who inherits them will develop the disorder.
Scientists
have found rare genes that directly cause Alzheimer’s in only
a few hundred extended families worldwide. When Alzheimer’s
disease is caused by deterministic genes, it is called "familial
Alzheimer’s disease." True familial Alzheimer’s accounts
for less than 5 percent of cases.
Some cases
of early-onset AD, called familial AD (FAD), are inherited.
FAD is caused by a number of different gene mutations on chromosomes
21, 14, and 1, and each of these mutations causes abnormal proteins
to be formed. Mutations on chromosome 21 cause the formation
of abnormal amyloid precursor protein (APP) -- Alzheimer
disease type 1; A mutation on chromosome 14 causes abnormal
presenilin 1(PSEN1) to be made --Alzheimers disease
type 3, and; mutation on chromosome 1 leads to abnormal presenilin
2 (PSEN2)--Alzheimer disease type 4. Since kindred's with
autosomal dominant EOFAD with no identifiable mutations in PSEN1,
PSEN2, or APP have been described; thus, it is
likely that mutations in other genes are causative. Molecular
genetic testing for PSEN1, PSEN2, and APP is available in clinical
laboratories.
How
does the APOE-4 gene affect Alzheimer's risk?
APOE
is 299 amino acids long and transports lipoproteins, fat-soluble
vitamins, and cholesterol into the lymph system and then into
the blood. It is synthesized principally in the liver, but has
also been found in other tissues such as the brain, kidneys,
and spleen. In the nervous system, non-neuronal cell types,
most notably astroglia and microglia, are the primary producers
of APOE, while neurons preferentially express the receptors
for APOE. The APOE gene, ApoE, is mapped to chromosome 19. The
gene is polymorphic[4] with three major alleles, ApoE2, ApoE3,
ApoE4, which translate into three isoforms of the protein: normal
- ApoE-3; dysfunctional - ApoE-2 and ApoE-4. These isoforms
differ from each other only by single amino acid substitutions
at positions 112 and 158,[5].
The
E4 variant is the only unequivocal genetic risk factor for late-onset
Alzheimer's disease in a variety of ethnic groups. Caucasian
and Japanese carriers of 2 E4 alleles have between 10 and 30
times the risk of developing AD by 75 years of age, as compared
to those not carrying any E4 alleles.
Apolipoprotein
E4 Potentiates Amyloid ? Peptide-induced Lysosomal Leakage and
Apoptosis in Neuronal Cells*
..."These
findings are consistent with apoE4 forming a reactive molecular
intermediate that avidly binds phospholipid and may insert into
the lysosomal membrane, destabilizing it and causing lysosomal
leakage and apoptosis in response to A?1–42".
Apolipoprotein
E fragments present in Alzheimer's disease brains induce neurofibrillary
tangle-like intracellular inclusions in neurons.
..."These
results suggest that apoE4 preferentially undergoes intracellular
processing, creating a bioactive fragment that interacts with
cytoskeletal components and induces NFT-like inclusions containing
phosphorylated tau and phosphorylated neurofilaments of high
molecular weight in neurons".
Apolipoprotein
Receptor 2 and X11{alpha}/ß Mediate Apolipoprotein E-Induced
Endocytosis of Amyloid-ß Precursor Protein and ß-Secretase,
Leading to Amyloid-ß Production
ApoE4
(along with other apolipoproteins) attaches itself to a receptor
on the surface of brain cells. That receptor, in turn, adheres
to a protein known as amyloid precursor protein. The entire
protein mass is then transported inside the cell where cutting
enzymes – called proteases – attack the amyloid precursor protein.
Additional
Genetic Factors
Researchers
in the U.K. and France have found three genes that make Alzheimer's
disease more likely when certain mutations are present. The
genes -- which are called CLU, CR1, and PICALM -- may make good
targets for new Alzheimer's disease treatments. Source: Genome-wide
association study identifies variants at CLU and PICALM associated
with Alzheimer's disease
CLU
and Picalm genes account for 9 percent of cases each, and the
CR1 gene is responsible for 4 percent. If we were able to remove
the detrimental effects of these genes through treatments, we
could reduce the proportion of people developing Alzheimer’s
by 20 percent,” said Julie Williams, the study’s lead author
and a professor of neuropsychological genetics at Cardiff University
in Wales.
The CLU
gene produces clusterin, which may protect the brain against
damage from pathogens or kick in to calm an inflammatory response.
Others seem to escort out excess amyloid, the plaque that builds
up in patients’ brains.
A second
study
published in Nature Genetics, by Philippe Amouyel from Institut
Pasteur de Lille in France, pinpointed CLU and CR1.
Currently
available drugs for Alzheimers
Four medications
are currently approved by regulatory agencies such as the US
Food and Drug Administration (FDA) and the European Medicines
Agency (EMEA) to treat the cognitive manifestations of AD: three
are acetylcholinesterase inhibitors and the other is memantine,
an NMDA receptor antagonist. No drug has an indication for delaying
or halting the progression of the disease.
|
Drug
Name
|
Molecular
Structure
|
Mechanism
of Action
|
Use
|
| Aricept®
(generic name: donepezil) Razadyne®, formerly known as Reminyl
(generic name: galantamine) |
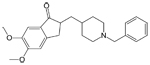 |
acetylcholinesterase
inhibitor --Prevents the breakdown of acetylcholine in the
brain |
For
people with mild ,moderate or severe AD |
| Exelon®
(generic name: rivastigmine) |
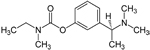 |
acetylcholinesterase
inhibitor --Prevents the breakdown of acetylcholine and
butyrylcholine (a brain chemical similar to acetylcholine)
in the brain |
For
people with mild or moderate AD |
| Razadyne®,
formerly known as Reminyl (generic name: galantamine) |
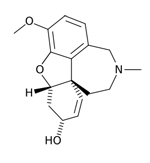 |
acetylcholinesterase
inhibitors--Prevents the breakdown of acetylcholine and
stimulates nicotinic receptors to release more acetylcholine
in the brain |
For
people with mild or moderate AD |
| Memantine
--Memantine is marketed under the brands Axura and Akatinol
by Merz, Namenda by Forest, Ebixa and Abixa by Lundbeck
and Memox by Unipharm |
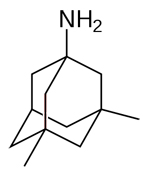 |
acting
on the glutamatergic system by blocking NMDA glutamate receptors
--Blocks the toxic effects associated with excess glutamate
and regulates glutamate activation |
Used
to treat moderate to severe AD |
Note: Cognex
(generic name: tacrine) also an acetylcholinesterase inhibitors
is not commonly used because of a number of side effects.
Cholinesterase
inhibitors are the most widely used drugs for Alzheimer's disease.
Cholinesterase inhibitors stop the breakdown of acetylcholine,
a chemical in the brain used for memory and other mental functions.
These types of medications help increase the levels of acetylcholine.
In Alzheimer’s disease, there is a deficiency in acetlycholine
in some areas of the brain, which accounts for some of the symptoms
of the disease.
It is important
to remember that these medications only slow the progression
of dementia and Alzheimer's disease – they do not stop or reverse
their course. These medications typically help for only months
to a few years and may not work as well once the disease progresses.
In general, individuals who use cholinesterase inhibitors experience
few side effects. The most commonly-experienced side effects
are gastrointestinal problems, such as nausea, diarrhea, vomiting,
and loss of appetite.
Source:
http://www.nia.nih.gov/Alzheimers/Publications/medicationsfs.htm
Research
Strategies
Intervention
strategies Researchers in Alzheimer's disease have identified
several strategies as possible interventions against amyloid:
Beta-Secretase
inhibitors. These work to block the first cleavage of APP
outside of the cell. *
Gamma-Secretase
inhibitors (e. g. Semagacestat). These work to block
the second cleavage of APP in the cell membrane and would then
stop the subsequent formation of Aß and its toxic fragments.
Selective
Aß42 lowering agents (e. g. Tarenflurbil). These modulate
gamma-secretase to reduce Aß42 production in favor of other
(shorter) Aß versions. *
Immunotherapies.
These stimulate the host immune system to recognize and attack
Aß or provide antibodies that either prevent plaque deposition
or enhance clearance of plaques.
Anti-aggregation
agents.These prevent Aß
fragments from aggregating or clear aggregates once they are
formed. There is some indication that supplementation of the
hormone melatonin may be effective against amyloid.
______________
Inhibition
and Reversal of Tau Fibrils:
Researchers
from
UC Santa Barbara are investigating a water-soluble extract
of cassia cinnamon that contains a class of small organic molecules
that inhibit the aggregation of tau and disassembles fibers
that have already formed, suggesting that neurofibrillary tangles
can possibly be reversed by these compounds. The extract exhibits
potent inhibitory activity, is orally available, water-soluble,
non-toxic, and the bioactive molecules are likely brain permeable.
The extract is readily produced in large quantities and can
be encapsulated in powder form for oral administration.
TauRx
Therapeutics
from Singapore and Allon Therapeutics of Vancouver, presented
human trial data for drugs that target tau. In a second-stage
trial presented at the conference, one of the doses of the TauRx
drug that was tested was able to dramatically slow the progression
of the disease; the effect was almost twice as big as typically
seen with existing drugs, Wischik says. "We have stopped the
progression of AD for 19 months," he claimed. The drug tested
is an old chemical that previously has been used for a variety
of purposes, including treating urinary tract infections.
ATPZ
analogues:
These are drug-like inhibitors of AD tau protein clumping, as
reported in the Journal
Biochemistry---A number of ATPZ analogues were synthesized,
and structure-activity relationships were defined. Further characterization
of representative ATPZ compounds showed they do not interfere
with tau-mediated MT assembly, and they are significantly more
effective at preventing the fibrillization of tau than the Abeta(1-42)
peptide which forms AD senile plaques. Thus, the ATPZ molecules
described here represent a novel class of tau assembly inhibitors
that merit further development for testing in animal models
of AD-like tau pathology.
Antraquinones
inhibit Tau Fibrils
-- e.g., emodin which is also a cytotoxic anticancer drug.
ALCAR (acetyl-l-carnitine
HCL) in phase III clinical trails, manufactured by Sigma-Tau
Pharmaceuticals, attempts to provide a possible protective effect
against neuritic tangles.
Drugs
in the Pipeline for Alzheimers
A
variety of clinical research trials are underway with agents
that try either to decrease the amount of Aß1-42 produced or
increase the amount of Aß1-42 removed. It is hoped that such
therapies may slow down the rate of progression of Alzheimer's
disease.
| |
Molecular
Structure |
Mechanism
of Action |
Clinical
Trials |
| Bapineuzumab--Elan
and Wyeth |
This
is a Monoclonal Antibody |
Bapineuzumab
is an antibody to the beta-amyloid plaques
Note:
phase II trial, which found that bapineuzumab failed
to improve cognitive function in a test of 234 Alzheimer’s
patients after 18 months of treatment. |
PIII--Bapineuzumab
in Patients With Mild to Moderate Alzheimer's Disease
(ApoE4 Non-Carrier) -- Estimated Completion Dec.
2010
2014
: Bapineuzumab
did not improve clinical outcomes in patients
with Alzheimer's disease, despite treatment differences
in biomarkers observed in APOE e4 carriers |
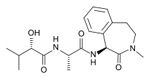
| Semagacestat
LY451039
-- Elli Lilly |
|
Gamma
secretase inhibitor-- These work to block the second
cleavage of APP in the cell membrane and would then
stop the subsequent formation of amyloid --Semagacestat
blocks the enzyme gamma-secretase which is responsible
for APP proteolysis |
Phase
III --Effect of Gamma-Secretase Inhibition on
the Progression of Alzheimer's Disease: LY450139 Versus
Placebo
in
August 2010, a disappointing interim analysis, in
which semagacestat performed worse than the placebo,
led to the trials being stopped |
| Solanezumab
(Eli
Lilly) |
This
is a Monoclonal Antibody |
Solanezumab
is a monoclonal antibody that binds specifically to
soluble amyloid beta and thereby alters the aggregating
characteristics of this peptide. |
Phase
III Data set to be released in 2012.. see
current study
Solanezumab,
a humanized monoclonal antibody that binds amyloid,
failed
to improve cognition or functional ability. |
| Gantenerumab |
This
is a Monoclonal Antibody |
Gantenerumab
is a monoclonal antibody that is currently being evaluated
in a prodromal Alzheimer's Disease population |
Gantenerumab: a novel human anti-Aß
antibody demonstrates sustained cerebral amyloid-ß
binding and elicits cell-mediated removal of human amyloid-ß.
See
2012 study.. see
current study |
| Dimebon
-latrepirdine --from Medivation |
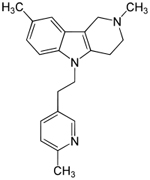 |
This
drug is an antihistamine used for 25 years in Russia--- |
Phase
III- A Phase 3 Efficacy Study Of Dimebon In Patients
With Moderate To Severe Alzheimer's Disease --Estimated
Study Completion Date: July 2011 |
| Flurizan
(*) Myriad
generic
name tarenflurbil --“enantiomer,” or mirror-image
molecule, of the non-steroidal anti-inflammatory drug
flurbiprofen |
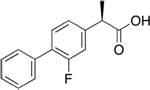 |
Lowers toxic Aß42 production by selectively modulating,
but not inhibiting, gamma-secretase activity to shift
cleavage of amyloid precursor protein (APP) away from
Aß42 production toward shorter, less toxic peptide fragments.
This drug failed a PIII trial. |
Fails
PIII -- See
Another Alzheimer’s Drug Fails in Large-Scale Trials |
| rember™--
Tau Aggregation Inhibitor (First and Second Generation)
-- TauRx |
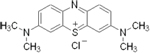 |
blocks the formation of Tau oligomers -- ability to
dissolve the tau fibers -- |
Phase
II completed.TRx0014
in Patients With Mild or Moderate Alzheimer's Disease
Phase
II - Ongoing -Open
Label Study of TRx0014 in Alzheimer's Disease |
| PBT2
8-Hydroxyquinoline
derivative |
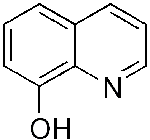 Note:
Image of 8-Hydroxyquinoline
Note:
Image of 8-Hydroxyquinoline |
targets
metal-induced aggregation of Aß, |
Completed
a Phase
IIa study in early Alzheimer's Disease patients
and has demonstrated safety and tolerability and showed
improvement in executive function
Plans
for PBT2 to Advance to Phase IIb
|
Note:
Why did Flurizan fail? In the past several years, evidence has
mounted that amyloid-beta-42, long considered the culprit in
the disease, affects memory-related functions only when it has
formed multi-protein conglomerations called “oligomers.” In
the light of this concept, it is possible that Flurizan affects
amyloid-beta-42 production in the brain and reduces the formation
of insoluble amyloid deposits but has little or no effect
on amyloid oligomer levels.
Initially,
it was thought that the insoluble amyloid plaques were the pathologic
culprits in AD. However, emerging evidence implicates soluble
Aß aggregates as the mediators of neurotoxicity. The Aß rapidly
aggregates by two separate pathways. The first leads to soluble
oligomers, referred to as Aß-derived diffusible ligands (ADDLs),
referred to as ADDLs. In a separate pathway, monomers can also
form protofibrils that eventually generate fibrillar aggregates
that coalesce into the characteristic insoluble amyloid. Several
lines of in vivo evidence suggest that ADDLs and protofibrils
, rather than monomeric Aß or insoluble amyloid plaques, mediate
neurotoxicity.
LATEST
NEWS UPDATES
Update July 31, 2017- Dementia: BACE inhibitor improves brain function
The protein amyloid beta is believed to be the major cause of Alzheimer's disease. Substances that reduce the production of amyloid beta, such as BACE inhibitors, are therefore promising candidates for new drug treatments. Scientists have recently demonstrated that one such BACE inhibitor reduces the amount of amyloid beta in the brain. By doing so, it can restore the normal function of nerve cells and significantly improve memory performance. read more
Update January 25, 2017 --Drug compound halts Alzheimer's Related Damage
Now, a new drug that could treat Alzheimer's disease, SAK3, has been developed by a Japanese research group led by Tohoku University Professor Kohji Fukunaga.In their study, the researchers found that the T-type calcium channel enhancer, SAK3, stimulates the release of acetylcholine in the brain and improves cognition by activating the memory molecule CaMKII. read more...
Update July 25, 2016 --Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer’s disease
..."We show that prolonged shifts in gut microbial composition and diversity induced by long-term broad-spectrum combinatorial antibiotic treatment regime decreases Aβ plaque deposition...These findings suggest the gut microbiota community diversity can regulate host innate immunity mechanisms that impact Aβ amyloidosis.. Abstract and Article
Update March 18, 2016 --Tryptophan-2,3-dioxygenase (TDO) inhibition ameliorates neurodegeneration by modulation of kynurenine pathway metabolites
Genetic inhibition of two KP enzymes—kynurenine-3-monooxygenase and tryptophan-2,3-dioxygenase (TDO)—improved neurodegeneration and other disease symptoms in fruit fly models of AD, PD, and HD, and that alterations in levels of neuroactive KP metabolites likely underlie the beneficial effects. Furthermore, it was found that inhibition of TDO using a drug-like compound reverses several disease phenotypes, underscoring the therapeutic promise of targeting this pathway in neurodegenerative disease... See Article in PNAS).
Update November 14, 2015 --Experimental drug J147 targeting Alzheimer's disease shows anti-aging effects
The Salk team expanded upon their previous development of a drug candidate, called J147 (see information and molecular structure of J147) , which takes a different tack by targeting Alzheimer's major risk factor–old age. In the new work, the team showed that the drug candidate worked well in a mouse model of aging not typically used in Alzheimer's research. When these mice were treated with J147, they had better memory and cognition, healthier blood vessels in the brain and other improved physiological features, as detailed. Salk Institute says Human Trials to begin in 2016. Journal Aging November 12, 2015 in the See previous research-- Abstract.. Full Article...The neurotrophic compound J147 reverses cognitive impairment in aged Alzheimer's disease mice --
Update July 20, 2015 --Novel antibodies show promise for Alzheimer's disease treatment--
NeuroPhage's NPT088 --Universally Targets Misfolded Proteins in Preclinical Studies Highlighted in Oral Session at the Alzheimer's Association International Conference-- (Fusion Protein -- antibody--) -- See The Virus that could cure Alzheimers.
See also: Scientists at NYU have evidence that monoclonal antibodies they developed may provide the blueprint for effective treatments for Alzheimer's disease and other neurodegenerative diseases, such as Parkinson's disease. Read news release
Update June 11,2015 --Axovant goes publc --main product candidate, RVT-101, aims to treat dementia in patients with Alzheimer’s disease --
RVT-101 has demonstrated statistically significant results on cognition as measured by the ADAS-cog and on function as measured by the ADCS-ADL in a randomized, placebo-controlled 684-subject study spanning 48 weeks of therapy, administered once-daily on a background of stable donepezil therapy. RVT-101 was well-tolerated by subjects in all 13 clinical trials conducted by GSK. We intend to commence a confirmatory phase 3 study in 2015.
Update Sept 11,2014 --In mouse
model of Alzheimer's disease, targeted immune booster removes
toxic proteins
Alzheimer's
disease experts at NYU Langone Medical Center and elsewhere
are reporting success in specifically harnessing a mouse's
immune system to attack and remove the buildup of toxic
proteins in the brain that are markers of the deadly neurodegenerative
disease. Researchers say the immune booster reduced both
amyloid beta plaques and tau tangles. Clinical Trial could
begin in 2015...
see full text of news release see Abstract
of study.
Update August 22, 2014 - Creating
pomegranate drug to stem Alzheimer's, Parkinson's
Research
will look to produce compound derivatives of punicalagin
for a drug that would treat neuro-inflammation and slow
down the progression of Alzheimer's disease, scientists
report. see Abstract
in Molecular Nutrition
New Update Jan 2014 --Dominantly
Inherited Alzheimer Network Trial: An Opportunity to Prevent
Dementia (DIAN TU)
This international study will
assess the safety, tolerability, and biomarker efficacy
of the drugs gantenerumab and solanezumab in individuals
who have a genetic mutation for autosomal-dominant Alzheimer's
disease. see full
text
New
Update -- June 18, 2013 ---Reversing the loss of brain connections
in Alzheimer’s disease -
A
New Drug --NitroMemantine-- Reverses Loss of Brain Connections
in Alzheimer's by shutting down hyperactive eNMDA receptors
on diseased neurons, NitroMemantine restores synapses between
those neurons...."The
Food and Drug Administration-approved drug memantine offers
some beneficial effect, but the improved eNMDAR antagonist
NitroMemantine completely ameliorates Aß-induced synaptic
loss, providing hope for disease-modifying intervention
in AD. "..- See
abstract in PNAS.
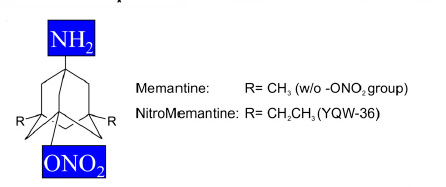
Molecular
structure for NitroMemantine ---- Source: Talantova
et al. 10.1073/pnas.1306832110
New
Update - March 26, 2012 --PLoS ONE Journal Publishes Mechanistic
Model of Alzheimer's Disease Endorsing Prana's PBT2 .
"This
paper builds on Prana’s previously published findings that as
we age our ability to maintain normal zinc distribution deteriorates.
Abeta forms amyloid by capturing and holding zinc, which in
turn further reduces our ability to maintain normal zinc distribution.
“This is a vicious pathological cycle. PBT2 interrupts this
cycle, re-distributing zinc needed for healthy brain function..”
See
Full Article:
The Zinc Dyshomeostasis Hypothesis of Alzheimer's Disease.
New
Update - March 13, 2012 Potential Alzheimer's Disease Drug Slows
Damage and Symptoms in Animal Model
A
compound that previously progressed to Phase II clinical trials
for cancer treatment slows neurological damage and improves
brain function in an animal model of Alzheimer's disease, according
to a new study. Summary
from ScienceDaily
The
Microtubule-Stabilizing Agent, Epothilone D, Reduces Axonal
Dysfunction, Neurotoxicity, Cognitive Deficits, and Alzheimer-Like
Pathology in an Interventional Study with Aged Tau Transgenic
Mice
New
Update - February 14, 2012 --Turmeric-Based Drug Effective On
Alzheimer Flies
"Curcumin,
a substance extracted from turmeric, prolongs life and enhances
activity of fruit flies with a nervous disorder similar to Alzheimers,
according to new research. The study conducted at Linköping
University, indicates that it is the initial stages of fibril
formation and fragments of the amyloid fibrils that are most
toxic to neurons..." Summary
from ScienceDaily
Source:
Curcumin
Promotes A-beta Fibrillation and Reduces Neurotoxicity in Transgenic
Drosophila
**New
Update February 9, 2012 --Alzheimer's Disease Symptoms Reversed
in Mice --The cancer drug bexarotene given to mice eliminates
brain-damaging proteins, leading to improved cognition.
The
cancer drug was given to mice and eliminated brain-damaging
proteins, leading to improved cognition within days, but it
is not known if it will work in humans. See review article in
Scientific American Online
**New
Update -- December 12, 2011 --The Journal of Biological Chemistry
published a study offering powerful validation of PBT2 as a
treatment for a number of neurodegenerative diseases, including
Alzheimer's, Parkinson's and Huntingtons diseases
PBT2
is Prana Biotechnology's lead drug for treating dementia in
Alzheimer's and Huntington's disease. It is a specific type
of 8-OHQ. Prana designed and selected PBT2 therapy for its enhanced
efficacy and tolerability.
See
the full publication: , "Different
8-OHQ's Protect Models of TDP-43, alpha-synuclein, and Polyglutamine
Proteotoxicity through Distinct Mechanisms".
***New
Update -- December 14, 2011 --Alzheimer's drug candidate may
be first to prevent disease progression Salk scientists develop
new drug that improves memory and prevents brain damage in mice
--
A
new drug candidate may be the first capable of halting the devastating
mental decline of Alzheimer's disease, based on the findings
of a study published today in PLoS one. When given to mice with
Alzheimer's, the drug, known as J147, improved memory
and prevented brain damage caused by the disease. The new compound,
developed by scientists at the Salk Institute for Biological
Studies, could be tested for treatment of the disease in humans
in the near future. "J147 enhances memory in both normal and
Alzheimer's mice and also protects the brain from the loss of
synaptic connections," says David Schubert, the head of Salk's
Cellular Neurobiology Laboratory, whose team developed the new
drug. "No drugs on the market for Alzheimer's have both of these
properties." Although it is yet unknown whether the compound
will prove safe and effective in humans, the Salk researchers'
say their results suggest the drug may hold potential for treatment
of people with Alzheimer's.
read more...
New
Update Novemember 29,2011 Surprisingly Few U.S. Physicians and
Payers Surveyed Are Familiar with Late-Stage Emerging Alzheimer's
Disease Therapies --
Resources, one of the world's leading research and advisory
firms for pharmaceutical and healthcare issues, finds that,
in the United States, surprisingly few surveyed neurologists
are familiar with key emerging therapies for Alzheimer's disease
on which they were surveyed.Less than one-quarter of surveyed
neurologists were familiar with the anti-beta-amyloid monoclonal
antibodies solanezumab (Eli Lilly) and bapineuzumab (Janssen
Alzheimer Immunotherapy/Pfizer) and less than 10 percent of
PCPs were familiar with these emerging disease-modifying drugs.
.. read
more
New
Update March 3, 2011 Scripps Research study points to
liver, not brain, as origin of Alzheimer's plaques
Unexpected
results from a Scripps Research Institute and ModGene, LLC study
could completely alter scientists' ideas about Alzheimer's disease—pointing
to the liver instead of the brain as the source of the "amyloid"
that deposits as brain plaques associated with this devastating
condition. The findings could offer a relatively simple approach
for Alzheimer's prevention and treatment. read
more
New
Update Jan 27, 2011 Stimulating The Brain's Immune Response
May Provide Treatment For Alzheimer's Disease
A
new target for the prevention of adverse immune responses identified
as factors in the development of Alzheimer's disease (AD) has
been discovered by researchers at the University of South Florida's
Department of Psychiatry and the Center of Excellence for Aging
and Brain Repair. read
more
New
Update Jan 6, 2011 -- Blood Test for Alzheimers
Using
a new technology that relies on thousands of synthetic molecules
to fish for disease-specific antibodies, researchers have developed
a potential method for detecting Alzheimer's disease with a
simple blood test. The same methodology might lead to blood
tests for many important diseases, according to the report in
the January 7th issue of the journal Cell, a Cell Press publication.
read
more
New
Update -- Jan 8 2010 --New approach to fighting Alzheimer's
shows potential in clinical trial Nutrient mix shows promise
in improving memory
In
a clinical trial of 225 Alzheimer's patients, researchers found
that a cocktail of three naturally occurring nutrients believed
to promote growth of those connections, known as synapses, plus
other ingredients (B vitamins, phosopholipids and antioxidants),
improved verbal memory in patients with mild Alzheimer's. --
read more
New
Update Oct. 16, 2009 -- IL-6 (Interleukin-6) shown to
remove plaque from Alzheimer's mouse model
Published
online in The FASEB Journal, data is based on the unexpected
finding that when the brain's immune cells (microglia) are activated
by the interleukin-6 protein (IL-6), they actually remove plaques
instead of causing them or making them worse. read
Abstract (Massive gliosis induced by interleukin-6 suppresses
A deposition in vivo: evidence against inflammation as a
driving force for amyloid deposition Paramita Chakrabarty, Karen
Jansen-West, Amanda Beccard, Carolina Ceballos-Diaz, Yona Levites,
Christophe Verbeeck, Abba C. Zubair, Dennis Dickson, Todd E.
Golde, and Pritam Das)
New
Update Jul 15, 2009 Significant Increase in Overall Executive
Function With PBT2
The
presentation was entitled "PBT2 ameliorates cognitive impairment
in Alzheimer's disease transgenic and aged mice: Evidence for
a common mechanism of action." New data indicates PBT2 benefits
not only Alzheimer's disease patients, but also could treat
the cognitive loss commonly associated with the normal ageing
process. PBT2 lowers amyloid burden in the brain and also corrects
metal imbalances that occur in the aged brain. Plans
for PBT2 to Advance to Phase IIb Clinical Trial Testing. See
News Release.
New
Update April 16, 2009 -- Depletion of circulating SAP
and , almost complete, disappearance of SAP from the CSF
Here,
in this unique study in Alzheimer's disease from the Journal
PNAS, the bis(d-proline) compound, (R)-1-[6-[(R)-2-carboxy-pyrrolidin-1-yl]-6-oxo-hexanoyl]pyrrolidine-2-carboxylic
acid (CPHPC), was shown to deplete circulating SAP and almost
complete, disappearance of SAP from the CSF. Kolstoe et.al.,
demonstrate that SAP depletion in vivo is caused by CPHPC cross-linking
pairs of SAP molecules in solution to form complexes that are
immediately cleared from the plasma. Article Title: Molecular
dissection of Alzheimer's disease neuropathology by depletion
of serum amyloid P component, See
Abstract.
Note:
Pentraxin Therapeutics Ltd, has acquired the full rights to
CPHC. In February 2009 Pentraxin Therapeutics Ltd licensed CPHPC
to GlaxoSmithKline for treatment of systemic amyloidosis, a
rare fatal disease.
References
1 - Shen
ZX (2004). "Brain cholinesterases: II. The molecular and cellular
basis of Alzheimer's disease". Med Hypotheses 63 (2): 308–21.
doi:10.1016/j.mehy.2004.02.031. PMID 15236795.
2 - a b
Wenk GL (2003). "Neuropathologic changes in Alzheimer's disease".
J Clin Psychiatry 64 Suppl 9: 7–10. PMID 12934968.
3 - Hardy
J, Allsop D (October 1991). "Amyloid deposition as the central
event in the aetiology of Alzheimer's disease". Trends Pharmacol.
Sci. 12 (10): 383–88. doi:10.1016/0165-6147(91)90609-V. PMID
1763432.
4- a b Mudher
A, Lovestone S (January 2002). "Alzheimer's disease-do tauists
and baptists finally shake hands?". Trends Neurosci. 25 (1):
22–26. doi:10.1016/S0166-2236(00)02031-2. PMID 11801334.
5 -Nistor
M, Don M, Parekh M, et al. (October 2007). "Alpha- and beta-secretase
activity as a function of age and beta-amyloid in Down syndrome
and normal brain". Neurobiol Aging 28 (10): 1493–1506. doi:10.1016/j.neurobiolaging.2006.06.023.
PMID 16904243.
6 - Lott
IT, Head E (March 2005). "Alzheimer disease and Down syndrome:
factors in pathogenesis". Neurobiol Aging 26 (3): 383–89. doi:10.1016/j.neurobiolaging.2004.08.005.
PMID 15639317.
43- Polvikoski
T, Sulkava R, Haltia M, et al. (November 1995). "Apolipoprotein
E, dementia, and cortical deposition of beta-amyloid protein".
N Engl J Med 333 (19): 1242–47. doi:10.1056/NEJM199511093331902.
PMID 7566000.
7- Transgenic
mice: Games D, Adams D, Alessandrini R, et al. (February 1995).
"Alzheimer-type neuropathology in transgenic mice overexpressing
V717F beta-amyloid precursor protein". Nature 373 (6514): 523–27.
doi:10.1038/373523a0. PMID 7845465.
Masliah
E, Sisk A, Mallory M, Mucke L, Schenk D, Games D (September
1996). "Comparison of neurodegenerative pathology in transgenic
mice overexpressing V717F beta-amyloid precursor protein and
Alzheimer's disease". J Neurosci 16 (18): 5795–811. PMID 8795633.
Hsiao K,
Chapman P, Nilsen S, et al. (October 1996). "Correlative memory
deficits, Abeta elevation, and amyloid plaques in transgenic
mice". Science (journal) 274 (5284): 99–102. doi:10.1126/science.274.5284.99.
PMID 8810256.
Lalonde
R, Dumont M, Staufenbiel M, Sturchler-Pierrat C, Strazielle
C. (2002). "Spatial learning, exploration, anxiety, and motor
coordination in female APP23 transgenic mice with the Swedish
mutation.". Brain Research (journal) 956: 36–44,year=2002. doi:10.1016/S0006-8993(02)03476-5.
PMID 12426044.
8 - Holmes
C, Boche D, Wilkinson D, et al. (July 2008). "Long-term effects
of Abeta42 immunisation in Alzheimer's disease: follow-up of
a randomised, placebo-controlled phase I trial". Lancet 372
(9634): 216–23. doi:10.1016/S0140-6736(08)61075-2. PMID 18640458.
^ Lauren J, Gimbel D, et al. (February 2009). "Cellular prion
protein mediates impairment of synaptic plasticity by amyloid-beta
oligomers". Nature 457 (7233): 1128-32. PMID 19242475.
9 Lauren
J, Gimbel D, et al. (February 2009). "Cellular prion protein
mediates impairment of synaptic plasticity by amyloid-beta oligomers".
Nature 457 (7233): 1128-32. PMID 19242475.
10- a b
Nikolaev, Anatoly; Todd McLaughlin, Dennis O'Leary, Marc Tessier-Lavigne
(19 February 2009). "N-APP
binds DR6 to cause axon pruning and neuron death via distinct
caspases". Nature 457 (7232): 981–989. doi:10.1038/nature07767.
ISSN 0028-0836. PMID 19225519.
11- Schmitz
C, Rutten BP, Pielen A, et al. (April 2004). "Hippocampal neuron
loss exceeds amyloid plaque load in a transgenic mouse model
of Alzheimer's disease". Am J Pathol 164 (4): 1495–1502. PMID
15039236.
12- Goedert
M, Spillantini MG, Crowther RA (July 1991). "Tau proteins and
neurofibrillary degeneration". Brain Pathol 1 (4): 279–86. doi:10.1111/j.1750-3639.1991.tb00671.x.
PMID 1669718.
13 - Iqbal
K, Alonso Adel C, Chen S, et al. (January 2005). "Tau pathology
in Alzheimer disease and other tauopathies". Biochim Biophys
Acta 1739 (2-3): 198–210. doi:10.1016/j.bbadis.2004.09.008.
PMID 15615638.
14 - Chun
W, Johnson GV (2007). "The role of tau phosphorylation and cleavage
in neuronal cell death". Front Biosci 12: 733–56. doi:10.2741/2097.
PMID 17127334.
15 - Itzhaki
RF, Wozniak MA (May 2008). "Herpes simplex virus type 1 in Alzheimer's
disease: the enemy within". J Alzheimers Dis 13 (4): 393–405.
ISSN 1387-2877. PMID 18487848. http://iospress.metapress.com/openurl.asp?genre=article&issn=1387-2877&volume=13&issue=4&spage=393.
16 - Moan
R (July 20, 2009). "MRI software accurately IDs preclinical
Alzheimer’s disease". Diagnostic Imaging. http://www.diagnosticimaging.com/news/display/article/113619/1428344.
|
