 |
Processing
of the amyloid precursor protein amyloid beta (A4) precursor
protein (peptidase nexin-II, Alzheimer disease) |
Amyloid beta
(Aß or Abeta) is a peptide of 39–43 amino acids that appear to
be the main constituent of amyloid plaques in the brains of Alzheimer's
disease patients. Similar plaques appear in some variants of Lewy
body dementia and in inclusion body myositis, a muscle disease.
Aß also forms aggregates coating cerebral blood vessels in cerebral
amyloid angiopathy. These plaques are composed of a tangle of
regularly ordered fibrillar aggregates called amyloid fibers,
a protein fold shared by other peptides such as prions associated
with protein misfolding diseases. Research on laboratory rats
suggest that the two-molecule, soluble form of the peptide is
a causative agent in the development of Alzheimer's and that the
two-molecule form is the smallest synaptotoxic species of soluble
amyloid beta oligomer [1] [2]
Formation
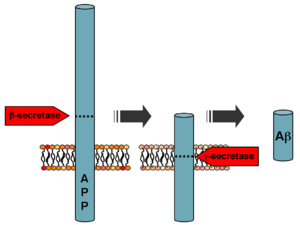
Aß is formed
after sequential cleavage of the amyloid
precursor protein, a transmembrane glycoprotein of undetermined
function. APP can be processed by a-, ß- and gamma-secretases;
Aß protein is generated by successive action of the ß and gamma-
secretases. The gamma secretase, which produces the C-terminal
end of the Aß peptide, cleaves within the transmembrane region
of APP and can generate a number of isoforms of 39-43 amino acid
residues in length. The most common isoforms are Aß40 and Aß42;
the shorter form is typically produced by cleavage that occurs
in the endoplasmic reticulum, while the longer form is produced
by cleavage in the trans-Golgi network.[3] The Aß40 form is the
more common of the two, but Aß42 is the more fibrillogenic and
is thus associated with disease states. Mutations in APP associated
with early-onset Alzheimer's have been noted to increase the relative
production of Aß42, and thus one suggested avenue of Alzheimer's
therapy involves modulating the activity of ß and gamma- secretases
to produce mainly Aß40.[4]
Genetics
Autosomal-dominant
mutations in APP cause hereditary early-onset Alzheimer's disease,
likely as a result of altered proteolytic processing. Increases
in either total Aß levels or the relative concentration of both
Aß40 and Aß42 (where the former is more concentrated in cerebrovascular
plaques and the latter in neuritic plaques)[5] have been implicated
in the pathogenesis of both familial and sporadic Alzheimer's
disease. Due to its more hydrophobic nature, the Aß42 is the most
amyloidogenic form of the peptide. However the central sequence
KLVFFAE is known to form amyloid on its own, and probably forms
the core of the fibril.
The "amyloid
hypothesis", that the plaques are responsible for the pathology
of Alzheimer's disease, is accepted by the majority of researchers
but is by no means conclusively established. Intra-cellular deposits
of tau protein are also seen in the
disease, and may also be implicated. The oligomers that form on
the amyloid pathway, rather than the mature fibrils, may be the
cytotoxic species.[6]
Intervention
strategies
Researchers
in Alzheimer's disease have identified five strategies as possible
interventions against amyloid:[7]
- Beta -Secretase
inhibitors. These work to block the first cleavage of APP outside
of the cell.
- Gamma Secretase
inhibitors (e. g. Semagacestat). These work to block the second cleavage
of APP in the cell membrane and would then stop the subsequent
formation of Aß and its toxic fragments.
- Selective
Aß42 lowering agents (e. g. Tarenflurbil). These modulate gamma-secretase to
reduce Aß42 production in favor of other (shorter)
Aß versions.
- Immunotherapies.
These stimulate the host immune system to recognize and attack
Aß or provide antibodies that either prevent plaque deposition
or enhance clearance of plaques.
- Anti-aggregation
agents[8].These
prevent Aß fragments from aggregating or clear aggregates once
they are formed.[9]
There is some
indication that supplementation of the hormone melatonin
may be effective against amyloid.[10][11]
Measuring
amyloid beta
There are
many different ways to measure Amyloid beta. One highly sensitive
method is ELISA which is an immuno-sandwich assay which utilizes
a pair of antibodies that recognize Amyloid beta.
Imaging compounds,
notable Pittsburgh Compound-B, (BTA-1, a thioflavin), can selectively
bind to amyloid beta in vitro and in vivo. This technique, combined
with PET imaging, has been used to image areas of plaque deposits
in Alzheimer's patients.
Atomic
force microscopy, which can visualize nanoscale molecular
surfaces, can be used to determine the aggregation state of Amyloid
beta in vitro.[12]
References
- Scmid, Randolf (June 2008). "New clue to Alzheimer's found" . Yahoo News. http://news.yahoo.com/s/ap/20080622/ap_on_sc/sci_alzheimer_s_clue.
- Shankar
GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I,
Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh
DM, Sabatini BL, Selkoe DJ (2008). "Amyloid-protein dimers
isolated directly from Alzheimer's brains impair synaptic
plasticity and memory". Nature Medicine 14 (June
22, 2008 online): 837.
- Hartmann
T, Bieger SC, Brühl B, et al. (1997). "Distinct sites
of intracellular production for Alzheimer's disease Aβ40/42
amyloid peptides". Nat. Med. 3 (9): 1016-20.
- Yin YI,
Bassit B, Zhu L, Yang X, Wang C, Li YM (2007). "γ-Secretase
Substrate Concentration Modulates the Aβ42/Aβ40 Ratio: Implications
for Alzheimer's disease". J. Biol. Chem. 282
(32): 23639-44.
- Lue LF, Kuo
YM, Roher AE, et al. (1999). "Soluble amyloid beta peptide concentration as a predictor of synaptic
change in Alzheimer's disease". Am. J. Pathol.
155 (3): 853-62.
- Kayed R,
Head E, Thompson JL, et al. (2003). "Common structure
of soluble amyloid oligomers implies common mechanism of pathogenesis".
Science (journal) 300 (5618): 486-9.
- Citron M (2004). "Strategies for disease
modification in Alzheimer's disease". Nat. Rev. Neurosci.
5 (9): 677-85.
- Lashuel
HA, Hartley DM, Balakhaneh D, Aggarwal A, Teichberg S, Callaway
DJE (2002). "New
class of inhibitors of amyloid-beta fibril formation. Implications
for the mechanism of pathogenesis in Alzheimer's disease".
J Biol Chem 277 (45): 42881-42890.
- Michael
H. Parker, Robert Chen, Kelly A. Conway, Daniel H. S. Lee;
Chi Luoi, Robert E. Boyd, Samuel O. Nortey, Tina M. Ross,
Malcolm K. Scott, Allen B. Reitz (2002). "Synthesis of (+)-5,8-Dihydroxy-3R-methyl-2R-(dipropylamino)-1,2,3,4-tetrahydro-naphthalene:
An Inhibitor of β-Amyloid1-42 Aggregation". Bioorg.
Med. Chem 10 (11): 3565-3569.
- Lahiri
DK, Chen DM, Lahiri P, Bondy S, Greig NH (November 2005).
"Amyloid, cholinesterase, melatonin, and metals and their roles
in aging and neurodegenerative diseases". Ann. N. Y.
Acad. Sci. 1056: 430-49.
- Wang
XC, Zhang YC, Chatterjie N, Grundke-Iqbal I, Iqbal K, Wang
JZ (June 2008). "Effect of melatonin and Melatonylvalpromide
on beta-amyloid and neurofilaments in N2a cells". Neurochem.
Res. 33 (6): 1138-44.
- Stine WB, Dahlgren
KN, Krafft GA, LaDu MJ (March 2003). "In vitro characterization
of conditions for amyloid-beta peptide oligomerization and
fibrillogenesis". J. Biol. Chem. 278 (13): 11612-22.
|
